首頁 ﹥ 動態消息 > 2018年媒體報導 > 醫療器械從研發到上市要花多少時間和金錢? 2018-11-06
▌一、概述
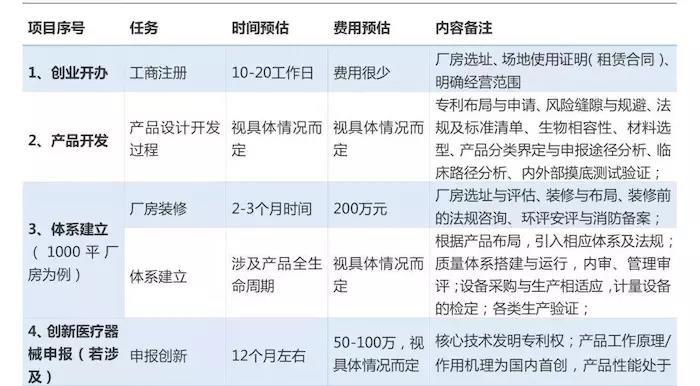
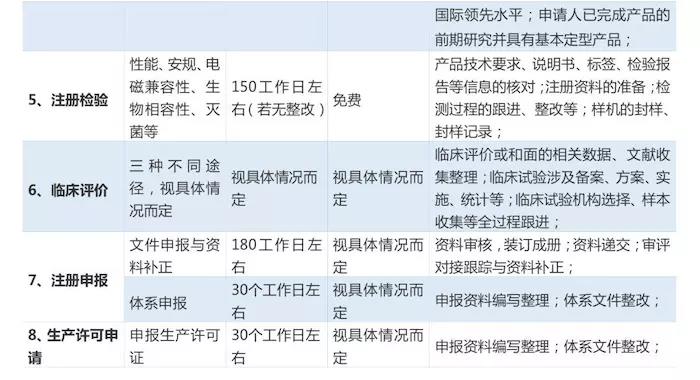
對於初創企業而言,產品從研發設計階段到走向消費市場,至少應經歷設計開發、註冊檢測、臨床試驗、註冊申報、生產許可申請等漫長的無盈利過程。那麼,如何評估產品安全性風險、申報註冊週期、投入資金與成本?都是初創企業創始人最關心的問題。
基於上述問題,筆者整理此文,希望對正踏步進軍醫療行業的初創企業有所幫助。本文將按照工商註冊、產品設計開發、體系建立、創新產品申報(若涉及)、註冊檢測、臨床試驗、註冊申報、生產許可申請、產品上市等必要階段進行簡單介紹。
▌二、基本流程
(一)
工商註冊
現如今,公司實行註冊資本認繳制和三證合一登記制度,註冊流程大為簡化,註冊費用大為節約,註冊週期也大為縮減。但在公司註冊前,需認真思考經營範圍,前置考慮未來產品生產範圍,避免後續再做變更。
(二)
產品開發設計
1、產品設計開發流程
產品設計開發可分7個階段:策劃階段、設計輸入階段、設計輸出階段、小試階段、中試階段、定型階段、註冊資料準備階段。詳情見圖1。
2、產品設計開發要點
一般而言,創始人創立公司是基於科研成果與產品佈局基礎上,並已初步具備研發團隊、合作夥伴等。但無論如何,對於初創公司而言,因涉及大量資金注入與技術攻堅,產品設計開發階段十分艱難,時間也是難以預估的,短則數月,長則數年。那麼,如何在產品設計開發階段降低成本,滿足需求,增加創新?
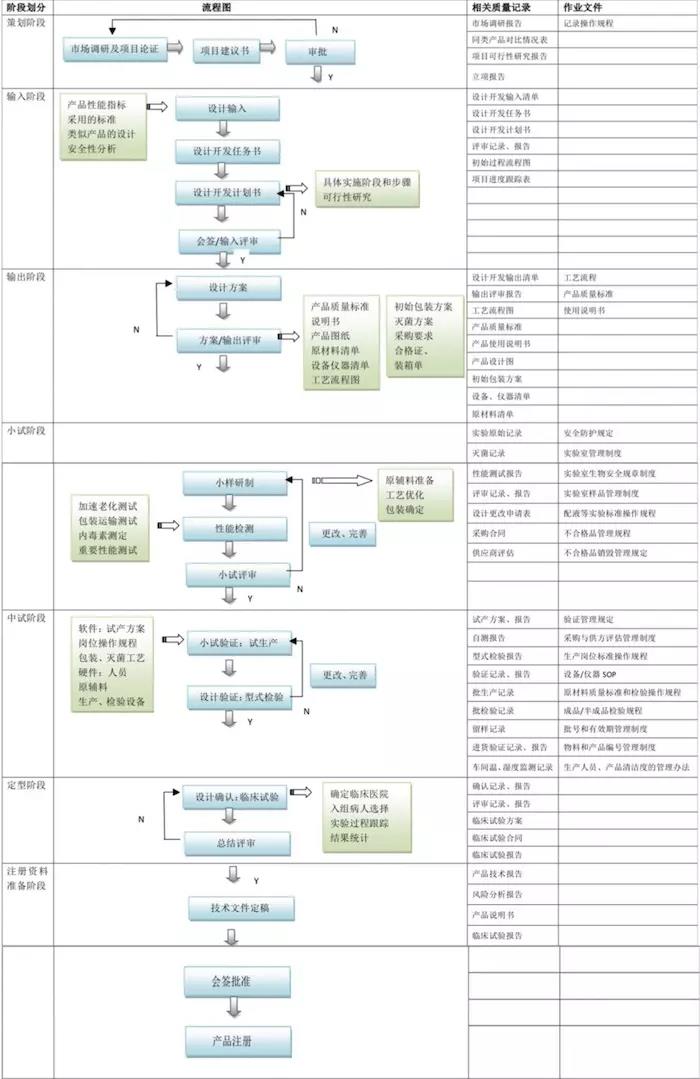
圖1.醫療器械產品設計開發流程圖
首先,產品技術層面,專業高效的研發團隊是前提和基礎;其次,法規層面,建議尋找專業CRO公司或引進法規團隊對產品開發進行嚴格的風險評估,明確產品申報類型等。但我們常常發現,很多新辦企業在產品已定型、開模後,才開始引進法規人員或尋找諮詢機構。此時的建議和措施往往基於補救,嚴重的可能涉及改模、重新設計等。所以對於新辦企業而言,法規前置慎重考量。最後,還應關注產品專利佈局。公司應重點佈局核心技術的智慧財產權保護,同時還應考慮核心技術轉讓、購買等問題,而產品的專利申報可以委託相應機構,後期若涉及專利較多,亦可引入兼職或專職人員。
3、創新醫療器械申報
2014年2月7日,CFDA發佈了《創新醫療器械特別審批程式(試行)》(食藥監械管〔2014〕13號),此文件自2014年3月1日起施行。該程式是在確保上市產品安全、有效的前提下,針對創新醫療器械設置的審批通道,但也有嚴苛的審批標準:(1)產品核心技術發明專利權。審批申請人經過其技術創新活動,在中國依法擁有產品核心技術發明專利權,或者依法通過受讓取得在中國發明專利權或其使用權;或者核心技術發明專利的申請已由國務院專利行政部門公開。(2)國內首創產品。主要工作原理/作用機理為國內首創,產品性能或者安全性與同類產品比較有根本性改進,技術上處於國際領先水準,並且具有顯著的臨床應用價值。(3)產品基本定型。申請人已完成產品的前期研究並具有基本定型產品,研究過程真實和受控,研究資料完整和可溯源。
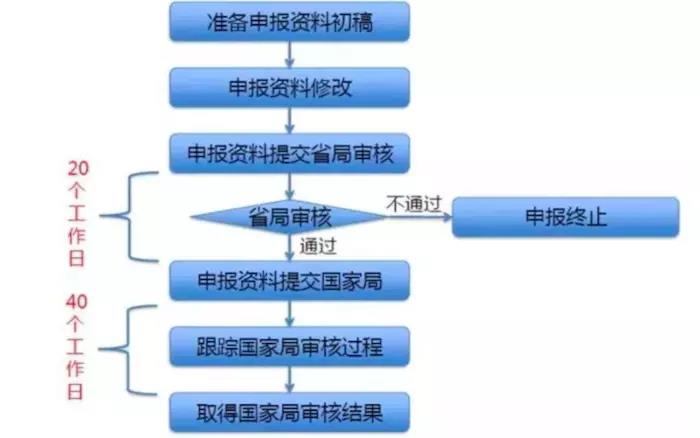
圖2.創新醫療器械申請流程
若企業產品設計滿足上述要求,則應著手申報創新。創新特別審批是申請人已完成產品的前期動物研究並具有基本定型產品即可申報,申報流程詳見圖2。
據統計,截止目前,CMDE共計收到創新醫療器械特別審批申請近800項(其中,進口產品40余項),通過審查一共149項(進口13項)。審查通過率僅約20%。因此,對於初創企業來說,建議尋找專業的CRO團隊合作,提高申報成功率。
(三)
體系建立
1、法規背景
為加強對醫療器械的監督管理,提升企業品質管制水準,保障醫療器械產品的安全有效,根據《醫療器械監督管理條例》(國務院令650號)和《醫療器械生產監督管理辦法》(國家食品藥品監督管理總局令第7號),國家食品藥品監督管理總局制定並印發了《關於發佈醫療器械生產品質管制規範附錄無菌醫療器械的公告》(2015年第101號)、《關於發佈醫療器械生產品質管制規範附錄植入性醫療器械的公告》(2015年第102號)及《關於發佈醫療器械生產品質管制規範附錄體外診斷試劑的公告》(2015年第103號)。上述三個附錄是無菌醫療器械、植入性醫療器械及體外診斷試劑三類產品生產品質管制規範的特殊要求,於2015年10月1日起正式施行。
與此同時,2017年9月1日,CFDA又發佈《關於第一類、第二類醫療器械生產企業實施醫療器械生產品質管制規範有關工作的通知》(食藥監辦械監〔2017〕120號),文件強調自2018年1月1日起,所有醫療器械生產企業均應當符合《醫療器械生產品質管制規範》要求。
因此,醫療器械生產企業需嚴格按照法規要求建立品質管制體系。而對於初創新公司除了體系建立,還需要面臨廠房選址和設計、建設等問題。
2、廠房規劃
對於廠房問題,需要根據產品管理類別進行考量。非無菌產品要求相對較低。若為無菌、體外診斷類產品,則應嚴格按照法規和標準選址,遠離有污染的空氣和水(如遠離鐵離、碼頭、機場、交通要道以及散發大量粉塵、屠宰場、染料等),對廠房的設計和裝修,必須請專業的團隊和公司來設計和施工,如行政區、生活區和輔助區不得相互防礙影響,空氣潔淨級別不同的潔淨室(區)之間的靜壓差應大於5帕,潔淨室(區)與室外大氣的靜壓差應大於10帕,空氣潔淨度級別進行合理佈局,人流、物流走向應當合理,避免交叉污染,注意潔淨室的水池或地漏等。雖然委託專業公司負責,但整個過程,都需要專業體系人員進一步把關,避免整改,比如消防、環評等通不過等。
3、人員配置
建立體系問題,初創團隊到底需要多少人?哪些崗位可以兼任?
必備崗位人員:生產負責人1名、研發部負責人1名、品質負責人1名、專職檢驗員2名、總經理1名;可兼任崗位人員:管理者代表1名,可由品質負責人兼任;採購部負責人1名,可由研發老大兼任;銷售部負責人1名,可由產品經理或總經理兼任;行政部負責人1名,也可讓總經理兼任;內審員2名,持有內審資格證的即可兼任,但2人不得在同部門;生產人員1名以上;
如此算來十多個崗位,兼任下來,至少應有6人以上,《醫療器械生產品質管制規範》及相關附錄中規定:“企業應當配備與生產產品相適應的專業技術人員、管理人員和操作人員,具有相應的品質檢驗機構或者專職檢驗人員的要求。要求生產負責人與品質負責人不得兼任,專職檢驗員也不要兼任”,雖然明確了三個崗位不得兼任,但也明確提到,應配備相適應的人員,所以還是應當根據產品配備充足的人員,避免被開出不合格專案。
4、體系認知
對於初創團隊而言,我們應深知:首先,品質管制體系是個系統工作,要有系統的觀念和思維。檔及記錄僅僅是整個系統的一部分,一個子系統而已,此外還有關鍵的管理控制子系統,設計控制子系統,生產製造子系統。需要與公司的培訓系統,績效系統,行銷系統等等結合互動。基於此,品質管制體系對企業來說,是企業多年運行的標準化沉澱產物。其次,符合規範要求是醫療器械企業生存的底線。行業特殊性,即法規符合性。以往,只知道要符合法規,但具體如何符合法規及指導觀念往往都是比較模糊的。當前法規的目標是確保品質管制體系的有效性以持續生產安全有效的醫療器械產品。基於此,法規監管並不希望企業三天兩頭修改技術檔,工藝檔等,而是應保障在當前法規要求下,可持續生產安全有效醫療器械產品。若日常工作中就對法規要求有所思慮,那麼品質管制體系法規符合性相對是較好的。再者,實施品質管制體系的終極目的,不是為了一張認證證書,而是降低風險。對企業來說,若證書拿到就萬事大吉,那麼企業發展必定不會長遠。監管部門審核,監督,你緊張;協力廠商審核你也緊張;大客戶來驗廠,估計十之八九你還是會緊張的。與其這樣,還不如老老實實的做好基礎,做好系統管理。更容易達到實施體系的目的:預防為主,降低風險。最後,我想說,沒有繼承,何來沉澱,何來文化。風來了,你是否準備好了?
(四)
註冊檢驗
1、法規背景
《醫療器械分類界定管理辦法》明確規定,申請第二類、第三類醫療器械註冊,應當進行註冊檢驗。醫療器械檢驗機構應當依據產品技術要求對相關產品進行註冊檢驗。註冊檢驗樣品的生產應當符合醫療器械品質管制體系的相關要求,註冊檢驗合格的方可進行臨床試驗或者申請註冊。辦理第一類醫療器械備案的,備案人可以提交產品自檢報告。
2、註冊檢驗內容
醫療器械註冊檢驗時檢驗機構(必須是CFDA認可有資質的機構)會依據企業所提供給的醫療器械產品技術要求做相應的檢驗,檢驗內容主要包括安規性能檢驗,EMC電磁相容性檢驗(有源產品需要,無源產品不需要),生物相容性檢驗等。
3、檢測週期
2017年3月15日,經國務院批准,財政部發佈《關於清理規範一批行政事業收費有關政策的通知》(財稅[2017]20號)。通知明確:自2017年4月1日起,包括醫療器械產品檢驗費在內的多項事業性收費取消徵收,這裡包括醫療器械註冊檢驗收費。
據統計,CFDA認可的全國醫療器械檢驗機構共有53家,各醫療檢驗機構對這一政策事先並無充分預案,但又必須按時執行。政策貌似助力行業發展,但短期內卻給行業發展帶來諸多混亂。企業送檢就要排長隊,過去60個工作日即可完成的,現在200個工作日也未必能完成。有些機構只接受本省樣品,跨省不接受,因為取消收費以後,經費均由本省承擔,沒有充足的資源對外提供服務。當然,CFDA也逐漸意識到當下這種混亂局面,領導非常重視,並委派相關人員深入調研。
針對現狀,企業能做的就是在產品開發立項過程中引入專業法規人員把關,協助研發工程師在設計之處就能明確遵循的標準和法規要求,降低後期整改難度。並在送檢前的開發階段做好充分的驗證測試,順利提高檢測通過率。
4、發展預測
不難發現,中國的醫療器械註冊檢驗制度體系正在重構,並逐漸形成如下趨勢:首先,製造商應當注重構建自己的自檢體系。因為放鬆管制意味著製造商將承擔更多的主體責任;其次,官方的檢驗機構正在重新定位,明確自己新的職責,如繼續承擔監督抽驗的任務,承擔強制性標準實施任務等等;最後,協力廠商檢驗機構迎來新機遇。協力廠商檢驗機構一直想在註冊檢驗領域為企業提供服務。過去沒有機會,現在機會出現了,大量協力廠商檢測機構未來會有很多的機會參與到整個檢驗體系的構建當中,分得一杯羹。檢驗資源增加了,企業會有更多的自主選擇權,這是好事。未來檢驗體系的改革一定是有利於產業發展的。
(五)
臨床評價
1、法規背景
《醫療器械註冊管理辦法》(國家食品藥品監督管理總局令第4號)
《醫療器械臨床評價技術指導原則》(2015年第14號通告)
《醫療器械臨床試驗品質管制規範》(國家食品藥品監督管理總局/中華人民共和國國家衛生和計劃生育委員會令第25號)
《關於醫療器械臨床試驗備案有關事宜的公告》(2015年第87號)
《免於進行臨床試驗的第二類醫療器械目錄》(2014年第12號通告)——488個II類產品
《免於進行臨床試驗的第三類醫療器械目錄》(2014年第13號通告)——79個III類產品
《第二批免於進行臨床試驗醫療器械目錄》(2016年第133號通告)——267個II類(含15個IVD),92個III類產品
《需進行臨床試驗審批的第三類醫療器械目錄》(2014年第14號的通告)——8個III類產品
《第三批免於進行臨床試驗醫療器械目錄》(2017年170號)——116個II類IVD,11個III類產品,37個II類產品
2、臨床評價路徑
臨床評價的三種途徑:對列入《免於進行臨床試驗的醫療器械目錄》中的產品,有條件的免於臨床試驗;對於同品種醫療器械臨床試驗或臨床使用獲得的資料進行分析評價;按照《醫療器械臨床試驗品質管制規範》開展臨床試驗;
3、實施建議
產品若不在目錄內,則只能通過臨床試驗或臨床評價兩個途徑。對於新公司首款產品,條件允許的話,建議做臨床試驗。首先,免臨床途徑很難拿到經驗資料和對比資料的授權;其次,首款產品註冊上市後,其他產品註冊也會很快啟動,便於後續產品臨床工作開展。關於臨床試驗工作,對於初創團隊而言,建議委託協力廠商有實力、專業的CRO團隊,利於更快、更好的推進臨床進度。
(六)
產品註冊申報
撰寫準備產品綜述資料、研究資料、生產製造資訊 、臨床評價資料、產品風險分析資料、產品技術要求、產品註冊檢驗報告、說明書和標籤樣稿等資料清單,整理遞交CFDA。
(七)
生產許可申請
1、法規背景
《醫療器械監督管理辦法》明確規定:從事第二類、第三類醫療器械生產的,生產企業應當向所在地省、自治區、直轄市人民政府食品藥品監督管理部門申請生產許可並提交其符合本條例規定條件的證明資料以及所生產醫療器械的註冊證。受理生產許可申請的食品藥品監督管理部門應當自受理之日起30個工作日內對申請資料進行審核,按照國務院食品藥品監督管理部門制定的醫療器械生產品質管制規範的要求進行核查。對符合規定條件的,准予許可並發給醫療器械生產許可證;對不符合規定條件的,不予許可並書面說明理由。
根據現行法規,醫療器械先註冊後許可。所以新辦企業會面臨一個特殊時期,拿到註冊證不能馬上銷售,需要申請“生產許可證”。以江蘇省為例,2014年12月22日,JSFDA發佈《關於明確江蘇省第二類、第三類醫療器械生產企業註冊與生產許可環節品質管制體系現場核查相關事宜的通知》(蘇食藥監械管〔2014〕369號),通知明確:為減少重複現場考核、提高工作效率,第二類和第三類醫療器械首次註冊的現場考核與生產許可環節品質管制體系現場考核,原則上合併進行。因此,對於新設企業首款產品而言,產品註冊證和生產許可證基本可以同步拿到。
2、申請週期
生產許可申請,法規規定為30個工作日,即1個月的時間。若現場審核無重大缺陷,並整改順利,基本上2-3個月即可拿到生產許可證。在此階段,公司可提前預熱,做好市場推廣,參展試用,但切記不可銷售。所以,在申請生產許可期間,公司基本上全部流程、檔都已形成,人員也已全部到位。萬事具備,只欠生產許可證。
3、生產許可證與註冊證差異
(1)醫療器械產品註冊證
醫療器械產品註冊證是醫療器械產品上市銷售的合格證明。第一類醫療器械產品備案,向市級食品藥品監督管理局提交備案資料。第二類醫療器械產品註冊,向所在地省、自治區、直轄市食品藥品監督管理部門提交註冊申請資料。第三類醫療器械產品註冊,註冊申請人應當向國家食品藥品監督管理局(CFDA)提交註冊申請資料。
(2)醫療器械生產許可證
醫療器械生產許可證是醫療器械生產企業獲得醫療器械產品生產的資質證明。從事第一類醫療器械生產的,向市食品藥品監督管理局備案。從事第二類、第三類醫療器械生產的,省、自治區、直轄市食品藥品監督管理局申請生產許可並提交相應的證明資料以及所生產醫療器械的註冊證。
▌三、費用預算與流程
產品註冊過程中,初創團隊最關心的就是費用及時間。以下為詳細流程表,涉及每個階段時限和費用。
(一)
費用
對於資金投入,並未嚴格細化。但可粗算下:工商註冊費用可忽略不計;研發投入需結合具體產品和團隊,難以預估。但以5人的研發團隊計算,人力資源成本約100萬/年。廠房建立涉及廠房裝修和後期體系建設。廠房裝修和租賃費用,以1000平米計算,裝修約200萬人民幣。租金按照30元/平米/月計算,一年就是36萬,加上水電物業將近50萬。品質體系建設是個系統工作,涉及產品的全生命週期,費用簡單預估50萬左右。註冊檢測不收費,若產品免臨床也不產生費用,做臨床的話根據產品而定,十幾萬至幾千萬都有可能。國產III類產品首次註冊費用為15.36萬,II類產品首次註冊費用則由各省級價格、財政部門制定。以江蘇省為例,II類產品首次註冊費用8.45萬元。小微企業提出的第二類醫療器械產品首次註冊申請,可免收首次註冊費。
(二)
時間
時間方面,我們也可簡單粗算:廠房選址按照3個月計算,公司註冊耗時30個工作日左右;廠房裝修需3個月左右;若產品研發結束,已基本定型,可直接啟動註冊檢驗工作。無不合格專案修復或整改補檢,可按照150個工作日計算;若產品免臨床則相對簡單,做臨床的話則需根據產品而定,3個月至一兩年都是有可能的。資料準備充分後遞交CFDA受理,技術審評中心排隊審評並補正,預計耗時180個工作日。審評通過即可開展體系考核,若無重大缺陷項目,30個工作日後即可取得產品註冊證。II類和III類產品耗時差異主要體現在臨床方面,註冊核對總和審評階段差異其實並不大。簡單算來,除去前期的準備階段,產品註冊耗時約:360工作日+臨床試驗時間。